- Mitochondrial trifunctional protein deficiency
-
Mitochondrial trifunctional protein deficiency Classification and external resources OMIM 609015 DiseasesDB 34111 eMedicine ped/1284 Mitochondrial trifunctional protein deficiency is an autosomal recessive fatty acid oxidation disorder[1] that prevents the body from converting certain fats to energy, particularly during periods without food . People with this disorder have inadequate levels of an enzyme that breaks down a certain group of fats called long-chain fatty acids.
Contents
Signs and symptoms
Signs and symptoms of mitochondrial trifunctional protein deficiency may begin during infancy or later in life. Features that occur during infancy include feeding difficulties, lack of energy (lethargy), low blood sugar (hypoglycemia), weak muscle tone (hypotonia), and liver problems. Infants with this disorder are also at high risk for serious heart problems, breathing difficulties, coma, and sudden death. Signs and symptoms of mitochondrial trifunctional protein deficiency that may begin after infancy include hypotonia, muscle pain, a breakdown of muscle tissue, and a loss of sensation in the extremities (peripheral neuropathy). Some patients with MTP deficiency show a protracted progressive course associated with myopathy, recurrent rhabdomyolysis, and sensorimotor axonal neuropathy. These patients tend to survive into adolescence and adulthood.
Genetics
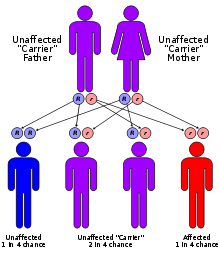 Mitochondrial trifunctional protein deficiency has an autosomal recessive pattern of inheritance.
Mitochondrial trifunctional protein deficiency has an autosomal recessive pattern of inheritance.
Mutations in the HADHA and HADHB genes cause mitochondrial trifunctional protein deficiency. These genes each provide instructions for making part of an enzyme complex called mitochondrial trifunctional protein. This enzyme complex functions in mitochondria, the energy-producing centers within cells. As the name suggests, mitochondrial trifunctional protein contains three enzymes that each perform a different function. This enzyme complex is required to break down (metabolize) a group of fats called long-chain fatty acids. Long-chain fatty acids are found in foods such as milk and certain oils. These fatty acids are stored in the body's fat tissues. Fatty acids are a major source of energy for the heart and muscles. During periods of fasting, fatty acids are also an important energy source for the liver and other tissues. Mutations in the HADHA or HADHB genes that cause mitochondrial trifunctional protein deficiency disrupt all three functions of this enzyme complex. Without enough of this enzyme complex, long-chain fatty acids from food and body fat cannot be metabolized and processed. As a result, these fatty acids are not converted to energy, which can lead to some features of this disorder, such as lethargy and hypoglycemia. Long-chain fatty acids or partially metabolized fatty acids may also build up and damage the liver, heart, and muscles. This abnormal buildup causes the other signs and symptoms of mitochondrial trifunctional protein deficiency.
Pathophysiology
The mitochondrial trifunctional protein, composed of 4 alpha and 4 beta subunits, catalyzes 3 steps in mitochondrial beta-oxidation of fatty acids: long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), long-chain enoyl-CoA hydratase, and long-chain thiolase activities. Trifunctional protein deficiency is characterized by decreased activity of all 3 enzymes. Clinically, classic trifunctional protein deficiency usually results in sudden unexplained infant death (SIDS; 272120), a Reye-like syndrome, cardiomyopathy, and/or skeletal myopathy.
Diagnosis
Onset of this disorder may begin during infancy or later in life. Signs and symptoms of the early onset form can include feeding difficulties, lack of energy (lethargy), low blood sugar (hypoglycemia), muscle weakness (hypotonia), liver problems, and a high risk for complications such as life-threatening heart and breathing problems, coma, and sudden unexpected death. The late-onset form is usually less severe; signs and symptoms can include hypotonia, muscle pain, a breakdown of muscle tissue, and abnormalities in the nervous system that affect arms and legs (peripheral neuropathy). Episodes of mitochondrial trifunctional protein deficiency can be triggered by periods of fasting or by illnesses such as viral infections. Diagnosis of this disorder is often confirmed using tandem mass spectrometry.[1]
Treatment
There is no known cure for this disease. Although the mortality rate among children with deficiency of LCHAD or complete deficiency of the trifunctional protein had been reported to be 75 to 90%, Ibdah et al. (1999) found that 67% of the affected children in their study were alive and receiving dietary treatment at the most recent follow-up, and most were able to attend school. Dietary treatment of children with fatty acid oxidation disorders dramatically reduced morbidity and mortality.
Epidemiology
Mitochondrial trifunctional protein deficiency is a rare disorder; its incidence is unknown.[citation needed]
External links
This article incorporates public domain text from The U.S. National Library of Medicine
- Mitochondrial trifunctional protein deficiency at NLM Genetics Home Reference
References
- ^ a b Solish JO, Singh RH (2002). "Management of fatty acid oxidation disorders: a survey of current treatment strategies". J Am Diet Assoc. 102 (12): 1800–1803. doi:10.1016/S0002-8223(02)90386-X. PMID 12487544.
- http://ghr.nlm.nih.gov/condition=mitochondrialtrifunctionalproteindeficiency#treatment
- http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=609015
- http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=600890
Inborn error of lipid metabolism: fatty-acid metabolism disorders (E71.3, 277.81–277.85) Synthesis Degradation Acyl transportGeneralMitochondrial trifunctional protein deficiency: Acute fatty liver of pregnancyOtherTo acetyl-CoASjögren–Larsson syndromeCategories:- Autosomal recessive disorders
- Fatty-acid metabolism disorders

Wikimedia Foundation. 2010.